来源: 日期:2024-10-12浏览:次
近日,广西医科大学第一附属医院河池医院·河池市人民医院肾内科接诊了一名患有遗传性罕见病——法布雷病的患者。
患者陈先生,今年42岁,3岁起便发现臀部、大腿内侧、背部有许多红色斑点,39岁起出现双下肢水肿,四肢出现烧灼般疼痛,到医院检查后发现尿蛋白高于正常值7倍多,并伴有反复四肢烧灼感、背部淤点、无汗,患者的母亲、外公均有上述症状,属于家族史,所以医生不排除是“法布雷病”。经基因检测后,患者被确诊为“法布雷病”。幸运的是,目前河池市人民医院已引入针对该罕见病的特效药,患者也已启动药物治疗。
(一)什么是法布雷病?
法布雷病是一种罕见的遗传性溶酶体贮积症,由编码a-半乳糖苷酶A(a-GalA)酶的GLA基因突变引起。a-GalA酶负责分解细胞内一种称为球三糖神经酰胺(Gb3)的脂肪物质。法布雷病的患病率约为1/40000至1/120000。
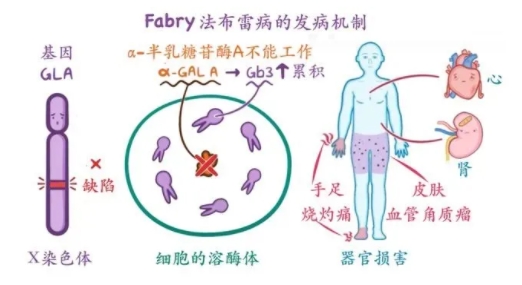
(二)有什么临床症状呢?
法布雷病作为一种罕见病,其初发症状特异性并不明显,且临床表现多样,全身多个器官和组织均可累及,在不同年龄、不同性别中存在很大差异。所以延误诊断率非常高,通常直至患者出现严重并发症(如慢性肾功能衰竭、心力衰竭、脑卒中)时才得以明确诊断。据报道,法布雷病儿童患者从首发症状到确诊平均延误约4年,成人患者15年,部分患者诊断延误可达30年或更长时间。症状包括:
皮肤表现:紫红色或深紫色斑点(血管角质瘤),通常出现在躯干和四肢。
肾脏问题:进行性肾功能衰竭,可能导致终末期肾病。
心脏问题:肥厚型心肌病,可导致心力衰竭、心律失常和猝死。
神经系统问题:疼痛、麻木、灼烧感和无力,通常影响手和脚。
消化系统问题:腹痛、腹泻和便秘。
眼部问题:角膜浑浊(角膜涡旋状浑浊)。
(三)遗传咨询和检查诊断
法布雷病是一种X连锁隐性遗传病,这意味着它主要影响男性。女性携带者通常没有症状,但可能会将疾病遗传给她们的儿子。
①如为男性患病,男性患者(父亲)将异常的X染色体传给女儿(发生率100%) ,患病的女儿为杂合子,临床表现相对轻,男性患者的儿子不会遗传此基因也不会发病。
②如为女性患病,女性患者(母亲)将异常X染色体传给子女的风险均为50%,男女患病概率均等,但患病的儿子为半合子,临床表现较明显。
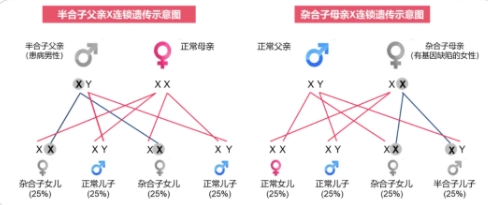
由此可见,对于有法布雷病家族史或其他遗传疾病风险的夫妇,建议在怀孕前进行遗传咨询,以讨论他们的选择和风险。对于已知携带相关基因突变的家庭,可以借助辅助生殖技术,如体外受精(IVF)结合胚胎植入前遗传诊断(PGT),可以在植入母体前筛查出携带基因突变的胚胎。
(四)法布雷病的诊断基于以下检查:
①病史和体格检查:寻找特征性症状和体征
②酶学检查:测量a-GalA酶活性
③基因检测:识别GLA基因突变
(五)治疗预后如何?
法布雷病的治疗旨在补充a-GalA酶,延缓或阻止疾病进展。治疗方法包括:酶替代疗法(ERT):定期静脉注射重组a-GalA酶底物减少疗法(SRT):使用药物米格司他汀抑制Gb3的合成。
法布雷病的预后取决于疾病的严重程度和治疗的及时性。早期诊断和治疗可以显著改善预后。如果不治疗,法布雷病患者的预期寿命可能会缩短。
 邮编:547000
邮编:547000
 总部院区地址:广西河池市金城江区金城中路455号
总部院区地址:广西河池市金城江区金城中路455号
中山院区地址:广西河池市金城江区中山路561号
备案号:桂ICP备07000541号-2  桂公网安备45120202451214号 联系电话:0778-2278191 COPYRIGHT © www.hchos.cn 广西河池市人民医院 版权所有 未经书面授权,不得转载、摘编、复制或者建立镜像
桂公网安备45120202451214号 联系电话:0778-2278191 COPYRIGHT © www.hchos.cn 广西河池市人民医院 版权所有 未经书面授权,不得转载、摘编、复制或者建立镜像
 急救电话:0778-2200000 0778-2299999
急救电话:0778-2200000 0778-2299999
